

Chemistry of Alzheimer's Disease
The Objectives of our Alzheimers Research
In 2000, we had realized that , logically, radicals generated by oxidative damage to peptides/proteins should underlie a number of disease states. We concentrated on the amyloid b-peptide (Ab) on which a literature consensus had developed as the causative agent of AD. Our research, as computational chemists, had two objective:1) to understand the chemical processes involved, and 2) to attempt design of small molecules that may prevent the disease.
Early ideas about the chemistry of Ab
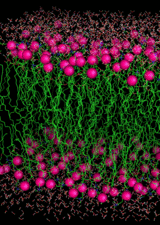
Membranes, Radicals, and Beta Sheets
A basic polyunsaturated membrane
The brain is very rich in polyunsaturated lipids. These are very susceptible to lipid peroxidation if a radical species can infiltrate to the hydrophobic interior of the bilayer. The brain uses a great deal of oxygen but has a low concentration of O2. Because O2 is more soluble in apolar media than water, the O2 concentrates in the interior of the bilayer. The amyloid beta peptide can damage neuronal membranes by lipid peroxidation and by generating non-ion-specific pores. The most toxic species is not the beta amyloid monomer but rather a soluble oligomeric form, dimer, trimer, etc. Lipid peroxidation requires an unsaturated membrane and an initialing radical that can penetrate hydrocarbon interior. The brain is rich in polyunsaturated lipids. The penetrating radical should be generated outside the membrane in an aqueous environment and be long-lived enough and hydrophobic enough to enter the bilayer. Recognizing that removal of the alpha-C hydrogen atom from any amino acid residue would generate a captodatively stabilized C-centred radical, we examined the possibilities of generating such a radical in a beta sheet, and which residues would be the most susceptible to alpha-C-H oxidation. The answer is a glycine residue in a beta sheet.
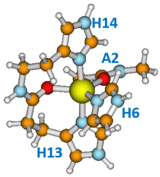
What about the Copper?
What is the affinity of Abeta for Cu(II) and Cu(I)?
Computations agree with experiment: for Cu(II)/Abeta, DG = -36 kJ/mol
The is some disagreement re binding of Cu(I): we calculate DG = -73 kJ/mol
How is the copper bound?
The graphic shows a model of the major binding motif. Cu(II) is bound to all three His residues, the carbonyl between H13 and H14, and the carbonyl of Ala2, in a trigonal bipyramidal arrangement. As many as 3 other pH-dependent structures have been proposed.
Cu(I) is tightly bound to His13 and His14 through the delta N atoms of the imidazole groups, in a linear dicoordinated configuration.
What is its reduction potential of Cu(II)/Abeta?
Computations agree with experiment: Eo(Cu(II)/Abeta, Cu(I)/Abeta) = 0.31 eV
The reduction is slow, requiring two higher energy intermediate structures.
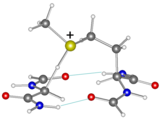
Is Met35 really involved?
Early literature says "YES"
Can it be oxidized by Abeta-bound Cu(II)?
Computations say "not directly". Either a Cu(I)-generated hydroxyl radical is involved, or a second Met. The latter suggests that at least a dimer must be involved.
Can the oxidizrd Met35 abstract a hydrogen atom from a Glycine backbone?
Computations say "not easily", and they must be close to each other in a beta sheet.
The graphic shows a model transition structure for H atom abstraction by Met(+) from a Gly in a beta sheet. The early calculations indicated a gas phase activation energy of 49 kJ.mol.
If glycine, which glycine?
Abeta ultimately oligomerizes as a beta sheet. We showed that in a beta sheet, only a glycine residue is susceptible to oxidation by H atom abstraction, and that an S-oxidized Met can effect the transformation. Of the six Gly residues, computations pointed to Gly29 and Gly33 as the most likely targets for oxidation on energetic grounds. Experimentally, the G33V mutant of Ab exhibits considerably reduced neurotoxicity (Kanski, J.; Varadarajan, S.; Aksenova, M. Butterfield, D. A. Biochim. Biophys. Acta, 2002, 1586, 190-198).
The Radical Model of Alzheimer's

The Radical Model of Alzheimer's
The Radical Model proposes that beta amyloid first aggregates into a soluble oligomer with antiparallel beta sheet structure and bearing a bound Cu(II). The amyloid-bound copper(ii) species generates a methionine sulfidyl radical either by direct electron transfer or indirectly by a hydroxyl radical. The methionyl radical in turn generates a captodatively stabilized alpha-C-centred radical at a glycyl residue, which can be long-lived enough to infiltrate into the interior of a polyunsaturated and oxygen-rich neuronal membrane. The membrane-insinuated alpha-C-centred glycyl radical then abstracts a H-arom to generate a more stable pentadienyl radical, which will react with oxygen to initiate lipid peroxidation. If the glycyl radical first intercepts an oxygen molecule before or after membrane insertion, the resultant peroxy species can also initiate lipid peroxidation by the same mechanism.
Those Neurotoxic Oligomers
Consensus is that the most neurotoxic species of beta amyloid are soluble aggregates (oligomers), rather than the monomeric form, or fibrular forms that make up the senile plaques that are one of the hallmarks of AD. While it is clear that amyloid fibrils possess a cross, in sync, antiparallel structure, oligomers appear to have an antiparallel beta sheet configuration. Toxicity has been shown to increase with extent of aggregation. Beyond that, little is known with certainty about the structures of oligomers, or their mechanism of toxicity.
The beta amyloid dimer (Ab(1-42)2)
Tjernberg showed that the site of Ab most likely to initiate aggregation is the central hydrophobic core, L17VFFA21. Various fragments of Ab containing this core aggregate as antparallel beta sheets and many ultimately form fibrils. We carried out long-duration (about 25 microsec in total), molecular dynamics simulations and found that the most stable conformation of the Ab(1-42) dimer is that shown at the right.
The most stable Ab(1-42) dimer
 dimer from MD simulations")
The most stable structure of the beta amyloid dimer derived from MD simulations. The blue and teal correspond to one strand, the red and pink to the other. The central region of the pink and teal is the LVFFA core in antiparallel configuration

Basic requirements


Example of SMD-US methodology Steered Molecular Dynamics is used to define the reaction coordinate. Then at selected points, a harmonic potential is applied and the system is equilibrated for 20 - 40 ns. Weighted histogram analysis (WHAM) is used to tease out the more accurate reaction coordinate and associated free energy changes.

Example of results from SMD-US methodology. Besides the black PMF curve with error bars, we also monitor H-bond count (blue curve), and separations of salt bridges (red and green curves).
Beta Sheet Inhibitors
Introduction
If oligomeric Ab is neurotoxic, and it aggregates as beta sheets, could one design molecules that can bind specifically to Ab and inhibit the beta sheet formation? That was the question that Samir Roy sought to answer in his PhD thesis research. At the time this work was initiated, Ab(1-42) was far too large and flexible to be studied by ab initio computational means, or even empirical MD simulations. As a model for full length Ab, we selected Ab(13-23), which, for short, we called Rec (for recognition site). This fragment encompasses the central hydrophobic core and includes three charged residues, K16, E22, and D23 which could be used to enhance binding affinity of the inhibitor. It also include two His residues, H13 and H14, which are the primary binding sites for Cu(II) and Cu(I). A strategic choice was to adopt a peptidic strand for the inhibitor as well, in order to maximize its selectivity for Rec (i.e., Ab) over the rest of the mish-mash of proteins that comprise a real biological system.
The design strategy
Having adopted a peptide platform for the inhibitor, the following requirements must be met:
- The length is 8 residues. Any larger and the immune system would destroy it.
- Use D-amino acids, and/or pseudopeptides to render it resistant to enzymatic degradation.
- Select residues of complimentary polarity: oppositely charged residues adjacent to each other and matching non-polar residues for the hydrophobic core. The oppositely charged residues may be used to direct the complexation to parallel or antiparallel beta sheets.
- Add methyl groups to the backbone to inhibit propagation of the beta sheet across the “inhibitor”
Four classes of inhibitor were designed by docking to Rec with the MOE semi-flexible docking software:
- SGA: L-residues which bind as an antiparallel beta sheet
- SGB: D-residues which bind as an antiparallel beta sheet
- SGC: L-residues which bind as a parallel beta sheet
- SGD: D-residues which bind as a parallel beta sheet
The methodology
The few best candidates from the MOE docking results in each class were subjected to MD simulations, using the GROMACS software, the Gromos96 53a5 forcefield, SPC water, in a 6 x 6 x 6 nm3 cube with periodic boundary conditions, at 1 atm pressure and 310 K. After first equilibrating the complex between Rec and the Inhibitor, steered molecular dynamics were applied to establish a reaction coordinate for dissociation by slowly pulling the two fragments apart to complete separation. Then 30 “windows” equally spaced along the path were selected and subjected to Umbrella Sampling. The difference between the high and low points along the potential of mean force curve were taken as the free energy difference between bound and unbound structures, i.e., the binding affinity, DGR-S. The procedure applied to the Rec dimer yielded a binding affinity, DGR-R = 53 kJ/mol. The inhibitor, S should bind maximally to R, i.e., DGR-S > DGR-R, and minimally to itself (DGS-S as small as possible).
A measure of effectiveness of inhibition is given by DGeff = 2 DGR-S – DGR-R – DGS-S
The results
Selected pseudopeptides (S) in each class are listed in the table below. A positive value for DGeff indicates that the peptide should be an effective beta sheet blocker (and prevent oligomerization of Ab).
Conclusions of the inhibitor study
The free energy changes in the table below correspond to stoichiometric amounts. One sees that SGB1 and SGC1 should make the best beta sheet inhibitors, although all classes may be effective if one can adjust the concentrations.
The ultimate challenge
Our last project was to modify SGC1 by adding a chelator with a high affinity for Cu(I). The modifies inhibitor we called TGC1. TGC1 has the additional property that the oxidation potential of its Cu(I) complex is too low to permit the generation of ROS. TGC1 should not only inhibit beta sheet formation but also remove any bound Cu from Abeta and prevent the formation of ROS. In the next section we describe this research.
| Peptide | Structure |
DGR-S kJ/mol |
DGS-S kJ/mol |
DGeff kJ/mol |
| SGA3 | N-Acetyl-Daba1-Orn2-MeLeu3-Phe4-MePhe5-Leu6-Ala7-Glu8-NH2 | 56 | 46 | 3 |
| SGB1 | N-Acetyl-daba1-orn2-leu3-mephe4-phe5-mephe6-leu7-glu8-NH2 | 62 | 45 | 26 |
| SGC1 | N-Acetyl-Glu1-Ala2-MePhe3-Phe4-MePhe5-Leu6-Orn7-Daba8-NH2 | 53 | 26 | 27 |
| SGD1 | N-Acetyl-glu1-leu2-mephe3-phe4-mephe5-leu6-orn7-daba8-NH2 | 50 | 32 | 15 |
where the lower case designates the D-amino acids and Daba = diaminobutyric acid, Orn = ornithine,
MeLeu = N-methylleucine, MePhe = N-methylphenylalanine. All these pseudo-peptides mimic the
R section of the Ab peptide; having complimentary charged residues between the hydrophobic
core. The all L-amino acid SGA and SGC bind to R in antiparallel and parallel mode, respectively.
The all-D-amino acid SGB and SGD bind to R in an antiparallel and parallel mode, respectively.
Data from: Banafsheh Mehrazma, Stanley Opare, Anahit Petoyan and Arvi Rauk, D-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors, Molecules 2018, 23, 2387-2410; doi:10.3390/molecules23092387
TGC1
Search for a Cu(I) chelator
Ab initio methods using the CAMB3LYP hybrid functional were applied to search for possible Cu(I)/Cu(II) chelators. The criterion was free energy of binding. Geometry and ZPVE and thermal corrections (change of enthalpy to 298K and entropy at 298 K, were determined with the 6-31+G(D) basis set with final zero point enthalpy calculated at the 6-311+G(2df,2p) level. Solvation was added by computation with the 6-31+G(d) basis set and SCRF = IPCM.
A variety of potential ligands, based on amines, thioethers, and imidazoles, were explored. The best ligand assembly, shown at the right involved a pyridine scaffold and two imidazoles forming a three-point chelation framework. For Cu(II), the fourth ligand was a water molecule. At the para position of the pyridine was placed a glutamic acid residue (the N-terminal residue of SGC1).
TGC1 - Rec Complexes, with and without Cu(I)
It remained to be proved that modification of SGC1 to TGC1, or attachment of Cu to Rec or to TGC1, did not disrupt or weaken its binding to Rec (and by extension, to Abeta). Indeed, equilibration by MD showed that all three combinations, TGC1-Rec, Cu(I)/Rec-TGC1, and Cu(I)/TGC1-Rec formed parallel beta sheets as expected. The Cu(I)/TGC1-Rec complex is shown at the right. The red backbone is that of Rec.
SMD_US PMF Analysis for TGC1-Rec-Cu(I)
The SMD_US procedures described above showed that the attachment of Cu(I) to either Rec or TGC1 enhanced the binding energy, which was greatest for Cu(I)/TGC1-Rec. The PMF curve is shown at the right,
Concluding Remarks
The latest (i.e. 2023) word on the chemistry of Alzheimer's disease supports the primary culpability of the beta amyloid peptide. The involvement of tau, the second hallmark of AD, is a downstream consequence of the buildup of amyloid aggregates. It confirms that we chose the correct direction 25 years ago when we entered the nascent AD field as computational chemists. I have described above, the two major accomplishment of our research, namely:
1) The discovery of a realistic structure of the Ab(1-42) dimer that initiates the oligomerization into more neurotoxic species, and
2) The development of a smallish molecular species, TGC1, that may simultaneously prevent the oligomerization and reduce copper toxicity.
It is my fondest hope that these ideas and results may be picked up by somebody that can develop them into affordable drugs that may prevent AD. In some of our publications and seminars we described our designer peptides as nanoantibodies. It was becoming clear that the first FDA approval of drugs against AD would be monoclonal antibodies. Three have emerged so far, aducanumab (Biogen, approved but controversial), lecanemab (Eisai/Biogen, approved but controversial) and donanemab (Lilly, approval pending)). It is also clear that these "mab" drugs would be prohibitively expensive for widespread use, hence the need for a "nanomab".
Optimum ligand assembly for Cu(II)/Cu(I) chelation, and properties

The final choice for Cu(I)/Cu(II) chelator. The binding affinities are sufficiently high for this complex to extract copper from Abeta. The reduction potential is low enough to prevent the reduction of oxygen to superoxide.
Complex between Cu/TGC1 and Rec

The complex between Cu-bound TGC1 and Rec
Dissociation PMF for Cu/TGC1-Rec

Binding of Cu/TGC1 to Rec
PREVENTION of AD
Familial Alzheimer's Disease (FAD)
About 5% of individuals have an unfortunate genetic predisposition to developing AD. For these individuals, the diagnosis of FAD may be as early as 40 years of age. Auguste D., the woman who first attracted Alzheimer's attention in 1901, had what would later be called FAD. As with the majority of AD cases (See below), the actual age of onset may be as many as 10 years before diagnosis. .Mutations in three genes are known to cause most cases of FAD. These are the presenilin 1 (PSEN1), presenilin 2 (PSEN2) and amyloid precursor protein (APP) genes. A mutation in PSEN1 is the most common of these three to cause FAD. Cleavage of APP results on the formation of Abeta. PSEN1 and PSEn2 are part of gamma-secretase, one of the enzymes responsible for the cleavage. All mutations result in a more rapid buildup of Abeta.
Age-related Alzheimer's Disease
The vast majority of individuals who contract AD do so from age 65 onward, the probability increasing with advanced age. As yet there is no cure, and with the extensive damage to the brain that has occurred before diagnosis, there may never be. However, early predictors of AD are becoming available, and it is hoped that administration of appropriate drugs will halt the progression before debilitating damage to the brain has occurred. Prior to the monoclonal antibody-based drugs mentioned above, approved drugs for AD (cholinesterase inhibitors: Donepezil , Rivastigmine, Galantamine; NMDA receptor antagonists: Memantine, Memantine+Donepezil) treat symptoms and, at best, result in a 6-18 month delay in the progression of disease symptoms. Even in the absence of pharmaceuticals, there are things that one can do to lower the probability of contracting AD.
Do we already have "drugs"?
Some over-the-counter or prescribed drugs have been tested for AD efficacy and shown to be beneficial:
NSAIDS - Ibuprofen (44%-60% reduction: NEJM 2001) - however,Ibuprofen (2009) and naproxen have been subjected to small clinical trials and shown to have no special benefits wrt AD prevention.
Statins - Simvastatin (≈ 50% reduction: Lancet, 2000) - large trial of simvastatin shows reduced risk (JAMA Neurol. 2017)
Coffee - Dark roast – NOT caffeine – phenylindoles (Nov 2018) - Oct 2019, in the CAIDE study, coffee drinking of 3-5 cups per day at midlife was associated with a decreased risk of dementia/AD by about 65% at late-life.
Vitamin D - Probably not (Jul 2018), but very low levels strongly increase risk of AD (Nov 2017)
Lifestyle Choices
Lifestyle choices that are deemed to be beneficial are:
Exercise - increases blood flow to the brain
Leisure activity and Social Engagement - lower stress (brain inflammation)
Mental Activity - "use it or lose it" increases brain plasticity (neuronal connections)
Sleep - allows clearance of debris, including amyloid plaques
Diet - intake of beneficial chemicals and avoidance of harmful ones
First Realization of AD

Alois Alzheimer (left) investigated a rare form of dementia in his patient, Auguste D. (right). After her death, he autopsied her brain and identified extracellular neuritic plaques and intracellular neurofibillary tangles. These are the hallmarks of what is now know as Alzheimer's disease. Auguste D had the early onset, or familial form.
Plaques and Tangles

This silver-stained image of an AD brain shows the hallmark plaques and tangles. The plaques were subsequently identified as deposits primarily consisting of the Abeta peptide. The tangles are aggregated of hyperphosphorylated tau protein.